ayotochtli
This project is maintained by sarbal
Figure 1
All data has been preprocessed and dumped into the data file. To run the analyses, please see the other notebooks referenced througout.
Load helper functions and data.
source("armadillo_helper.r")
source("load_data.r")
Panel C
Plot the collection timepoints for the five quadruplets.
plot(lubridate::dmy( timepoints$Received), timepoints$Quad , xlim=c(17167,17897), xlab="", ylab="", pch=19, col= candy_colors[as.numeric(timepoints$Quad)], cex=3)
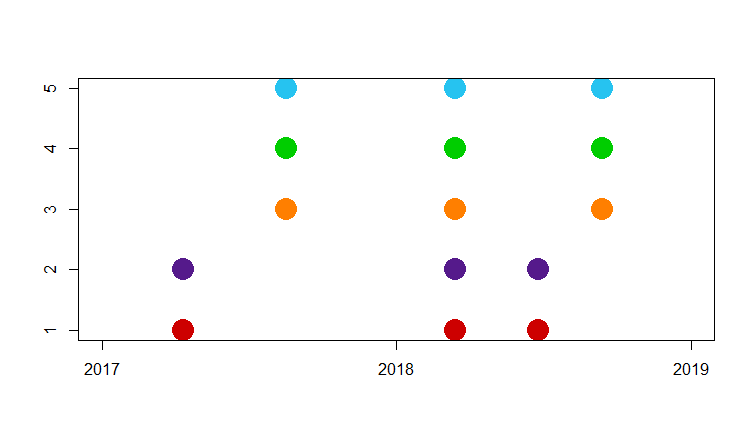
Panel D
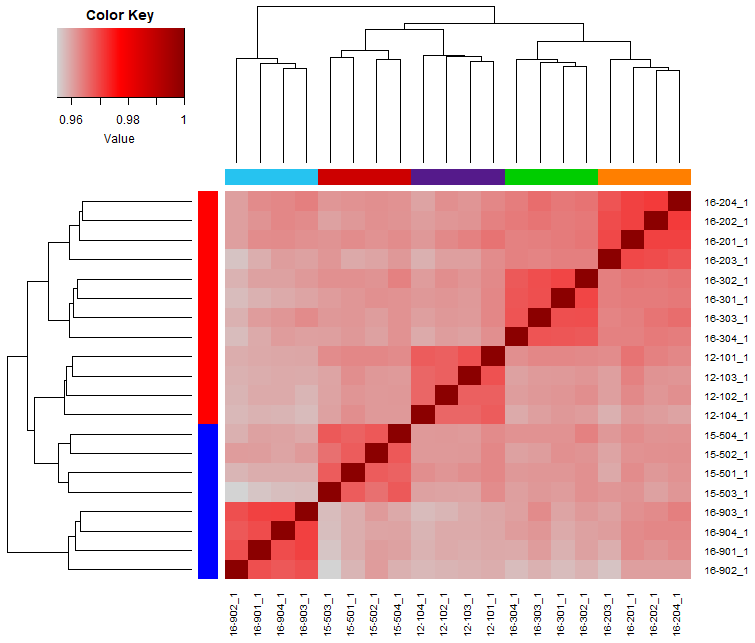
For all the timepoints, we use the CPM transformed RNA-seq expression data to calculate the sample similarity.
For the analyses on generating this data, see the RNA-seq notebook.
### Calculate sample-sample correlations
sample.cors = cor(X.cpm.all, method ="spearman", use="pair")
Then plot the first timepoint. r_samp has been defined in the armadillo_helper.r script.
### Plot heatmap of first timepoint
heatmap.3(sample.cors[r_samp,r_samp],
col=cols5,
ColSideCol = candy_colors[pData$Quad[r_samp]],
RowSideCol = sex_colors[pData$Sex[r_samp]])
## Repeated plot with different column labels
hm = heatmap.3(sample.cors[r_samp,r_samp],
col=cols5,
ColSideCol = candy_colors[pData$Quad[r_samp]],
RowSideCol = (candy_colors)[-(1:3)][pData$Lanes[r_samp]])
Panel F
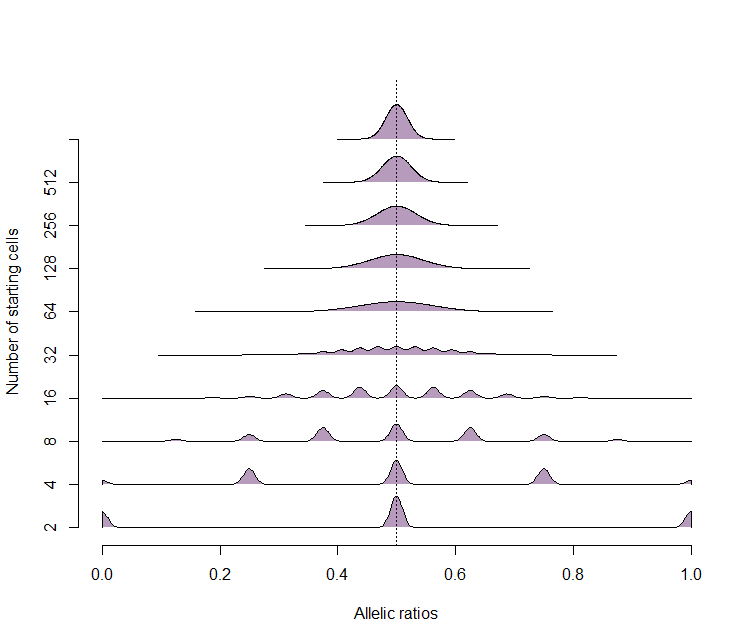
The model for allelic ratios based on starting number of cells is the binomial model Binom(n,p), with the probability of either allele at 0.5.
### Generate underlying data, selecting an allele based on number of starting cells
raw_data = lapply( 2^(1:10), function(i) rbinom(1e5, i, 0.5)/i )
names(raw_data) = 1:10
### Plot distributions for starting cell numbers
beanplot( raw_data, side="s", what = c(1,1,0,0), col=makeTransparent(viridis(10)), horizontal = T, bw = 0.01, cutmin = 0, cutmax = 1, frame.plot = F, boxwex = 2, axes=F, xlab="Allelic ratios", ylab="Number of starting cells"); axis(1); axis(2, at=1:10, labels= 2^(1:10))